Arq Bras Cardiol: Imagem cardiovasc. 2024; 37(3): e20240050
Is Wild-type Transthyretin Cardiac Amyloidosis Still Considered a Rare Disease?
DOI: 10.36660/abcimg.20240050i
Introduction
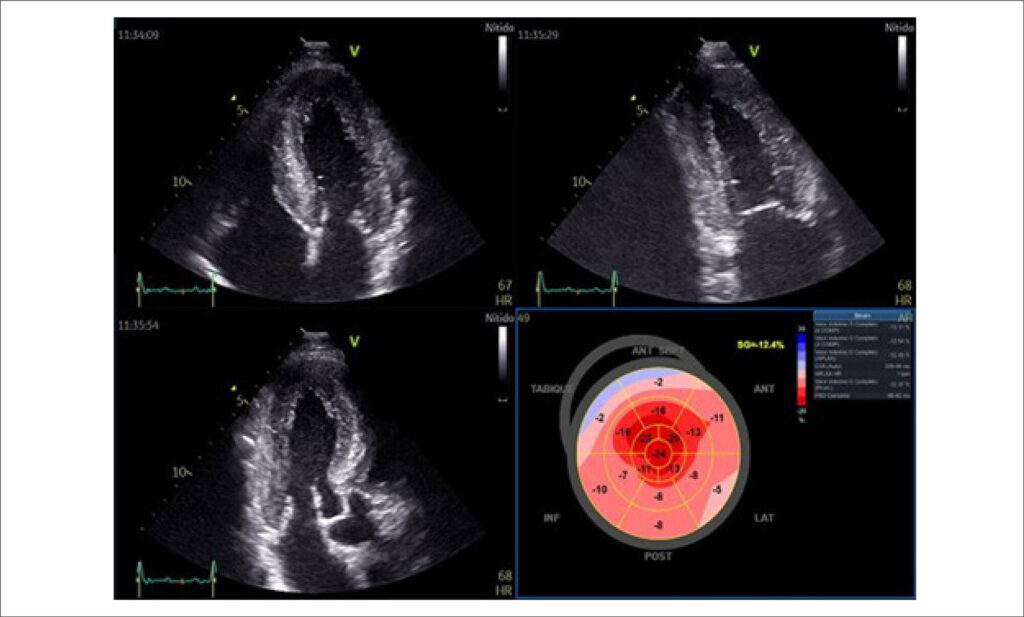
Amyloidosis is a disease characterized by extracellular deposition of insoluble proteins resulting in organ dysfunction. There are more than 30 known proteins capable of aggregating into amyloid fibrils; among these, amyloidosis due to light chain (AL) immunoglobulin and transthyretin amyloidosis (ATTR) represent 98% of cardiac amyloidosis. Transthyretin (TTR) is a tetrameric protein whose function is to transport thyroxine and retinol-binding protein. It is primarily produced in the liver, with smaller quantities found in the choroid plexus and retinal pigment epithelium.
There are two distinct forms of ATTR: hereditary, caused by pathogenic mutations that destabilize the protein, and acquired, also known as wild-type (wtATTR), which results from the accumulation of wtATTR protein. Regarding clinical manifestations of ATTR, in the case of the hereditary type, they depend on the genetic variant involved and can lead to cardiac and extracardiac involvement, including sensory-motor peripheral neuropathies, autonomic neuropathies, gastrointestinal manifestations, among others. In the case of wtATTR, cardiac involvement is the predominant manifestation, characterized by heart failure, conduction disorders, and arrhythmias. The gold standard for diagnosis of cardiac amyloidosis is the demonstration of apple-green birefringence in polarized light microscopy of Congo red-stained tissue. However, confirmatory biopsy is no longer necessary for a diagnosis when the following criteria are met: heart failure with an echocardiogram or cardiac magnetic resonance imaging consistent with amyloidosis, a grade 2 or 3 uptake on radionuclide scintigraphy with 99m-Technetium-labeled 3,3-diphosphono-1,2-propanodicarboxylic acid or pyrophosphate (PYP), and absence of a detectable monoclonal gammopathy. There are currently new disease-modifying therapeutic options available for both hereditary and acquired ATTR. Among the available drugs are selective TTR stabilizers, such as tafamidis, and genetic silencers, such as inotersen or patisiran, which provide the most significant benefit in the early stages of the disease.
[…]
Keywords: Amyloidosis; Prealbumin; Rare diseases
672