Arq Bras Cardiol: Imagem cardiovasc. 2025; 38(2): e20240091
My Approach to Diagnose Fabry Disease
DOI: 10.36660/abcimg.20240091i
Abstract
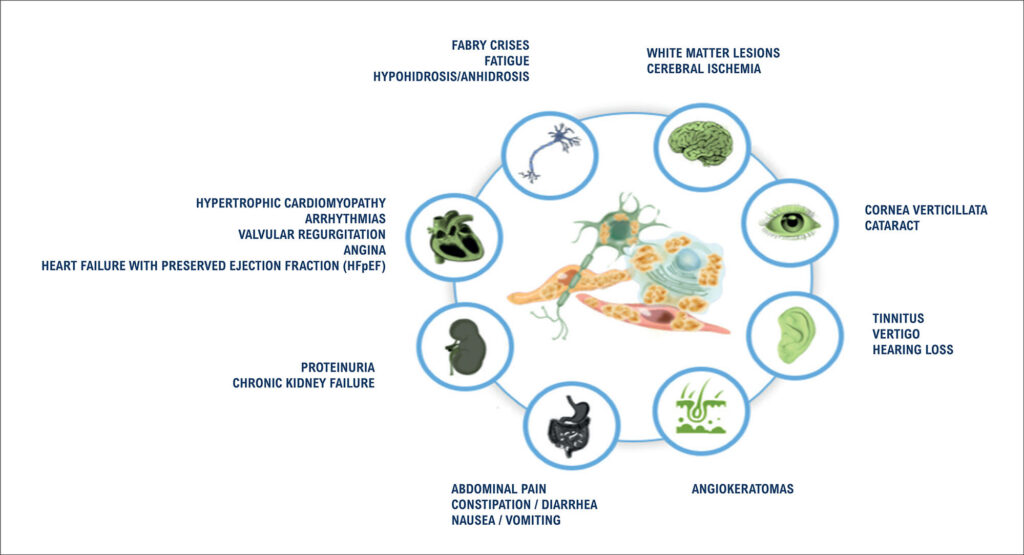
Fabry disease (FD) is an inherited lysosomal storage disorder caused by a deficiency of the enzyme alpha-galactosidase A (α-Gal A). This enzymatic defect leads to the cytoplasmic lysosomal accumulation of globotriaosylceramides (GB3 and LysoGB3), resulting in multisystemic clinical manifestations. Cardiovascular involvement, often mimicking hypertrophic cardiomyopathy, is the main determinant of morbidity and mortality, due to the development of arrhythmia, myocardial ischemia, and heart failure. Although FD is a rare condition in the general population, the availability of specific enzyme replacement therapy, which can alter the natural course of the disease, underscores the importance of including FD as a key differential diagnosis among storage cardiomyopathies. Diagnostic evaluation should encompass a thorough clinical assessment, with particular attention to patient history and physical examination, complemented by laboratory testing and imaging studies, such as electrocardiography and echocardiography. Cardiac magnetic resonance imaging, including late gadolinium enhancement and T1 and T2 parametric mapping, provides additional diagnostic and prognostic information and should ideally be performed at the time of initial diagnosis. Definitive diagnosis is established by genetic sequencing of the GLA gene, located on the long arm of the X chromosome, enabling the selection of the most appropriate therapeutic strategy for each patient.
861